|  |
Global Optimization | |
| |
Global Optimization | |
|
Minimization problems are frequently encountered in science and engineering. Unfortunately, such problems are hard to solve for systems with a large number of degrees of freedom. Often the number of minima grows at least exponentially with the number of degrees of freedom[i],[ii]. Most non-stochastic optimization algorithms can easily find one minimum, but usually it is not the global minimum[iii].
Figure 1 An illustration of protein structure. Only three amino acid monomers are shown.
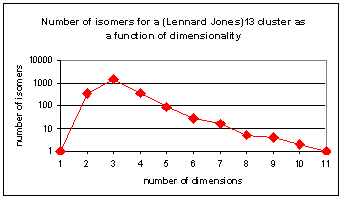
One very important global optimization problem is protein folding[iv]. A protein is a polymer made up of amino acids. (See Figure 1) Most amino acids have two easily rotatable bonds in the backbone, and many also have flexible side chains that add additional degrees of freedom to the protein. It is not computationally feasible to exhaustively sample all of a protein's configuration space, even if one considers only the torsional degrees of freedom. Many stochastic global minimization algorithms have been developed. They include genetic algorithms[v],[vi],[vii],[viii], Monte-Carlo[ix] based methods[x],[xi],[xii],[xiii], quantum annealing[xiv],[xv], and global optimization methods that modify the potential energy surface[xvi],[xvii]. Some of these involve artificially increasing the dimensionality of the system.ii It has been known for some time that the maximum number of minima of Lennard-Jones clusters occurs in three dimensions.[xviii],[xix] Figure 2 plots the number of minima of LJ13 as a function of the number of dimensions. As the number of dimensions is increased from three, the system gradually looses its complexity, until it has only one minimum in ten-dimensional space.
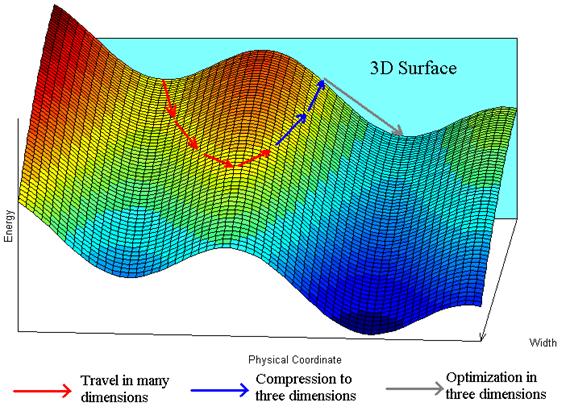
Figure 2 Number of known isomers for a (Lennard Jones)13 cluster as a function of dimensionality. LJ13 has the greatest number of isomers in 3 dimensions. Faken et al.[xx] developed one of the global algorithm that take advantage of extra dimensionality. Here are the steps of this algorithm. Suppose one wants to find the global minimum of a system described by a set of continuous coordinates R in D dimensions, where normally D = 3. 1. Minimize the potential energy V(R) in D dimensions using a non-stochastic algorithm such as steepest descent or conjugate gradient. The system arrives at a local minimum where V(R)=Elm. 2. Carry out a random walk at constant energy in D+H dimensions, where H ³ 1. At the beginning of the walk, the system is in D dimensions, and the extra coordinates are set to 0. At the end of the walk, the energy of the system is still equal to Elm, but the system is someplace in the D+H dimensional space. In the algorithm of Faken et al.xx all atoms are moved simultaneously. 3. Compress the system back to the D dimensional space, keeping the energy constant. 4. Optimize the geometry in D dimensions, setting extra coordinates equal to zero. The energy of the resulting minimum will be equal to or lower than that of the previous minimum (in 3D). 5. The procedure is either repeated by returning to step 2, or the current R is reported as the final answer.
Figure 3 Key steps in the algorithm of Faken et al.
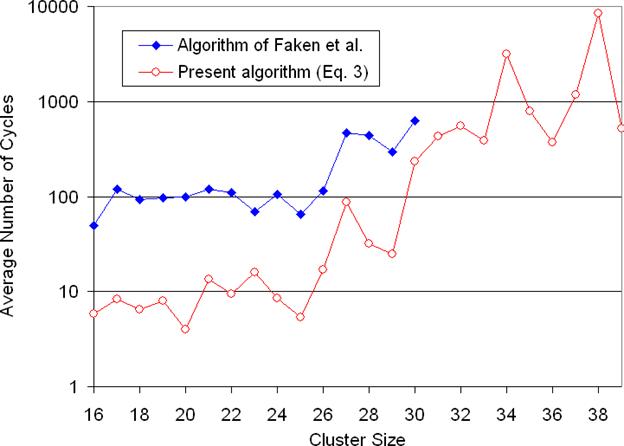
We have improved the speed of the algorithm of Faken et al.xx by three orders of magnitude. One order of magnitude improvement was achieved because our implementation of this global optimization algorithm required fewer cycles to find global minima. This is due to our improvements to both the 3D→4D walk at constant energy and to the 4D→3D compression step of the algorithm. Our implementation of the 3D→4D walk at constant energy differed from that of Faken et al.xx in two respects. 1) In our implementation the same number of steps was used in the 3D→4D walk at constant energy step for all clusters. 2) In cases where the compression step failed to bring the system to 3D, the system was returned to the previous local minimum in 3D. In the algorithm of Faken et al.xx it was brought back to the point in 4D from which the compression was started. Our test revealed that this change generally resulted in a small increase in efficiency in locating the global minimum in 3D. This makes sense, because overall the minima bear some degree of resemblance to each other, and taking the system further and further into 4D makes it look less like a 3D minimum. A much more significant acceleration of the algorithm of Faken et al.xx was achieved due to improvements made to the 4D→3D compression step. Faken et al.xx compressed the system from 4D to 3D at constant energy by moving atoms perpendicular to the energy gradient in the direction of decreasing width. We have been able to reduce the CPU time required for the compression step by two orders of magnitude by combining energy and width into a single function and minimizing that function using our own algorithm. Using this approach also increased the fraction of successful optimizations, because the domain of convergence for this method was greater than that for the approach of Faken et al.xx
Figure 4 Comparison of performance of our program with program of Faken et al. We optimized each Lennard-Jones cluster nine times, each time starting from a different random configuration. For LJ38 we have done twenty optimizations. We report the mean in the number of cycles needed to reach the global minimum.
[i] Garey, M. R.; Johnson, D. S. Computers and Intractability: A Guide to the Theory of NP-Completeness, (Freeman, San Francisco, 1979). [ii] Stillinger, F. H. Phys. Rev. E, 1999, 59, 48-51. [iii] Hugh M. Cartwright, Applications of Artificial Intelligence in Chemistry, (Oxford University Press, 1993). [iv] Dobson, C. M.; Fersht A. R. Protein Folding, Cambridge University Press, 1997. [v] Deaven, D. M.; Tit, N.; Morris, J. R.; Ho, K. M. Chem. Phys. Lett. 1995, 247, 339. [vi] Niesse, J. A.; Mayne, H. R.; J. Chem. Phys. 1996, 105, 4700. [vii] Gregurick, S. K.; Alexander, M. H.; Hartke, B. J. Chem. Phys. 1996, 104, 2684. [viii] Mesters, J.; Scuseria, G. E.; J. Comput. Chem. 1995, 16, 729. [ix] Metropolis, N.; Rosenbluth, A. W.; Rosenbluth, M. N.; Teller, A. H.; Teller, E. J. Chem. Phys. 1953, 21, 1087-1091. [x] Kirkpatrick, S.; Gelatt, C. D.; Vecchi, M. P. Science (Washington, D.C.) 1983, 220. [xi] C. J. Geyer, in Computing Science and Statistics: Proceedings of the 23rd Symposium on the Interface (American Statistical Association, New York, 1991), p. 156. [xii] Geyer, C. J.; Thompson, E. A. J. Am. Stat. Assoc. 1995, 90, 909. [xiii] Falcioni, M.; Deem, M.W. J. Chem. Phys. 1999, 110, 1754-1766. [xiv] Doll, J. D.; Freeman, D. L. IEEE Comput. Sci. Eng. 1994, 1, 22. [xv] Finnila, A. B.; Gomez, M. A.; Sebenik C.; Stenson, C; Doll, J. D. Chem. Phys. Lett. 1994, 219, 343. [xvi] Stillinger, F. H.; Stillinger D. K. J. Chem. Phys. 1990, 93, 6106. [xvii] Wales, D. J.; Doye, J. P. J. Phys. Chem. A 1997, 101, 5111-5116. [xviii] Doye, J. P. K.; Miller, M. A.; Wales, D. J. J Chem. Phys. 1999, 111, 8417-8428. [xix] Tsai, C. J.; Jordan, K. D. J. Phys. Chem. 1993, 97, 11227-11237. [xx] Faken, D. B.; Voter, A. F.; Freeman, D. L.; Doll, J. D. J. Phys. Chem. A, 1999, 103, 9521-9526. |